sma是什么疾病
sma是什么疾病

sma是什么疾病
脊髓性肌萎缩症 Spinal Muscular Atrophy 缩写:SMA是一组会导致肌肉无力和萎缩的运动神经元性疾病。运动神经起源于脊髓,控制着人体进行呼吸、爬、走、头颈控制以及吞咽等活动的肌肉。SMA是一种相对常见的“罕见病”——新生儿患病率为1:6000-1:10000。
SMA对人周身上下的肌肉都会造成侵害。在最常见的类型中,患者腿部无力的情况通常较手臂处更为严重。有时,吞咽和呼吸功能 比如呼吸、咳嗽、清除气道分泌物也会受到影响。当参与呼吸和咳嗽的肌肉受到侵害而变得无力时,就会加增患者罹患肺炎和呼吸道感染的风险,也会造成患者睡眠时呼吸困难。SMA患者的认知功能、感官系统不受影响。通常,基于发病时间及所能达到的最大运动功能,患者会被划分为4个类型 Ⅰ、Ⅱ、Ⅲ、Ⅳ。
sma的病因
SMA是由于人体内被称作为“运动神经元存活1号”基因 SMN1的缺失或异常 突变所导致的。对于健全人而言,这一基因可以制造出一种被称为“运动神经元存活” SMN的蛋白。若此基因异常的话,人体内的SMN蛋白就会缺失或显著减少,进而导致运动神经元出现严重的问题。运动神经元是位于脊髓内的神经细胞,由它们所延伸出来的神经纤维通向身体各处的肌肉。SMN蛋白对运动神经元的存活和健康至关重要,没有这种蛋白,神经细胞就有可能萎缩并最终死亡,从而导致肌肉的无力。随着SMA患儿的生长,他们无力的肌肉很难满足日常活动的需要。这样的肌肉无力还会导致骨骼和脊柱的变形,进而导致呼吸方面的问题和进一步的身体功能丧失。
依据患者的发病时间及所能达到的最大运动功能指标,SMA被分为Ⅰ、Ⅱ、Ⅲ、Ⅳ4个类型。但需要注意的是,每个SMA患者的病程可能都会有所差异。一般SMA不被视作为一种进行性疾病,但随着肌肉的不断弱化,患者典型的表现就是逐渐丧失 各种身体功能。这一过程可能是渐进式的,也可能在生长突增阶段或疾病期间更为明显。至于为何不同患者的肌肉弱化和身体功能丧失速度不尽相同,目前仍然难以解释。尽管通常的趋势是随着患者年龄增长,他们的身体功能会逐渐丧失,但研究人员也观察到有的患者在相当长的时间内,经常有数年之久,身体功能会保持稳定。
sma的遗传
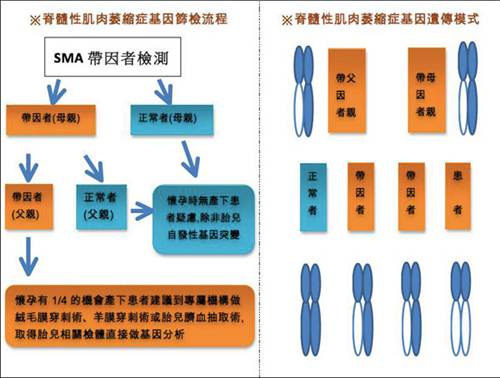
SMA是一种常染色体隐性遗传疾病。患儿父母通常各自只有一个SMN1基因有缺陷,所以并不感染SMA,也不会表现出患病的症状,他们被称作为携带者。当父母双方都是缺陷基因的携带者,并且各自把这个基因遗传给了孩子,才会导致孩子获得了两个有缺陷的基因拷贝而染病。
最常见的SMA是由5号染色体上的SMN1 Survival Motor Neuron运动神经元存活基因突变所引起,因此也被称作5q型SMA。
常规人群,约每50人中就有1个是SMA致病基因的携带者,父母同为携带者,他们每次怀孕都有:
l 25%的几率孩子会是SMA患者
l 50%的几率孩子会是SMA致病基因携带者
l 25%的几率孩子会是健康的
smaⅠ型
Ⅰ型SMA也被称作沃德尼格霍夫曼病 Werdnig-Hoffmann Disease。患儿通常在6个月内发病,头控能力较差,无法达到与发育相对称的运动功能。他们的典型特征就是无法独坐,需要依靠推车或轮椅。吞咽和喂养会变得困难,由于难以避免误吸 将分泌物或食物吸入肺内引起窒息,最后孩子会丧失安全吞咽的能力。最终,他们还可能需要借助饲管摄取流质食物来补充营养。不同的家庭对此有不同的看法,可以就是否使用饲管做出自己个人化的决定。若选择使用饲管,那么可能需要在孩子吞咽变得困难之前就进行放置,然而具体放置的时间也是一个个人化的选择。
Ⅰ型SMA患儿会出现呼吸肌 帮助扩张胸廓让肺内充满空气的肌肉无力的情况。他们的胸廓较正常情况要小,主要依靠腹部肌肉进行呼吸 腹式呼吸,因此肺部不能得到充分的发育,咳嗽会变得非常微弱。睡眠中肺内可能也难以吸入足够的空气,最终白天醒着时也会出现这样的问题。很多Ⅰ型患者会需要使用呼吸机来帮助呼吸,同样,这也取决于不同家庭各自的选择。肌肉无力还会导致脊柱的问题 侧弯和髋部的问题 脱臼,进而造成更多的身体功能丧失。较严重的脊柱侧弯可以使用支具 固定背心应对,有些情况则可以通过手术进行治疗。
smaⅡ型
Ⅱ型SMA患者通常在6-18个月发病。他们具有一系列的运动能力,但运动功能指标滞后。典型特征就是可以独坐,但有些孩子需要帮助才能进入坐姿。在支具或外力的辅助下,他们可能可以站立,但是无法行走,需要借助轮椅来移动。Ⅱ型SMA患儿通常不会出现吞咽方面的问题,但不同的孩子之间却会存在个体差异。有些患者可能无法自行摄入足够的食物以维持正常的体重和生长所需,这时若家人认为是最佳选择的话,可能就需要使用饲管来进行喂养。Ⅱ型患儿同样也可能出现呼吸肌无力,咳嗽困难的情况。其中一些可能需要在夜间使用呼吸机来帮助改善呼吸。肌肉无力同样会导致脊柱 侧弯和髋部 脱臼的问题,进而造成更多的身体功能丧失。较严重的脊柱侧弯可以使用支具 固定背心应对,有些情况则可以通过手术进行治疗。
smaⅢ型
Ⅲ型SMA通常也被称作库杰尔博格伟兰德病 Kugelberg-Welander或少年型脊髓性肌萎缩症。通常18个月后发病,时间更为宽泛,早至1岁,晚至青春期都可能发病。Ⅲ型SMA患者可以独自站立并行走,但随着病程的发展,某个时候行走就会出现困难。早期所应该达到的运动指标都正常,然而,一旦开始行走他们就会经常摔倒,俯身或坐在地上难以站起,可能也无法奔跑。Ⅲ型SMA患者手指伸出后会有细微的颤动,但舌部肌肉震颤的情况较罕见。喂食和吞咽的问题在他们童年期间并不常见。有些患者可能会在童年、青年甚至成年时丧失行走能力,但这经常都和他们身体的快速发育或其它疾病有关。
smaⅣ型 成年发病
成年型患者的症状通常始于35岁后。SMA很少于18-30岁之间发病。成年型SMA较其它类型要少见得多,被定义为要18岁后才出现无力的症状,但大多数报道的Ⅳ型病例都是在35岁后发病。其典型特征是发病隐匿,病情发展非常缓慢。延髓所控制的肌肉,即用于吞咽和呼吸的肌肉很少受到侵害。
SMA患者的身体功能通常会随着时间的推移而丧失,这在他们身体快速发育或患病期间显得较为突出,但基于目前的研究这种现象还无法解释。尽管随着年龄增长患者身体功能的丧失是普遍的趋势,但据观察他们在较长的时间段内 常会有数年之久身体功能会非常稳定。
宝,喜欢(sma是什么疾病 sma是什么病能活多久)记得收藏一下,以后访问不迷路。