基因组学一
基因组学一
Ø 基因
基因是1909年丹麦植物学家W.Johannsen根据希腊文单词genos(birth,给予生命)创造的。现代分子生物学的基因概念通常认为是:基因是储存和表达某一多肽链信息或RNA分子信息所必需的全部核苷酸序列(DNA序列),是生物体传递和表达遗传信息的基本单位。
Ø 基因组
基因组一词系由德国汉堡大学H.Winkles教授于1920年首创。最初是由GENs和chromosOME组成为“genome”,用于表示生物的全部基因和染色体组成的概念,是指生物的整套染色体所含有的全部DNA序列。现在一般认为,基因组即指生物所具有的携带遗传信息的遗传物质的总和,包括所有的基因和基因间区域。基因组的结构主要指核酸分子中不同的基因功能区域各自的分布和排列情况,其功能是储存及表达遗传信息。不同种类生物储存的遗传信息量迥异,其基因组的结构和组织形式也不同。
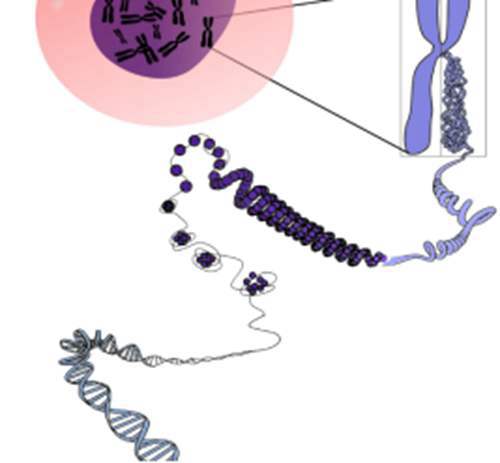
Ø 真核生物基因组
核基因组:细胞核内所有遗传物质的总和;
线粒体基因组:线粒体携带遗传物质的总和;
叶绿体基因组:叶绿体携带遗传物质的总和。
Ø 原核生物基因组
染色体:原核细胞内主要的遗传物质;
质粒:是独立于细菌染色体的自主复制的环状双链DNA分子。能稳定地独立存在于染色体外,并传递到子代,一般不整合到宿主染色体上,通常编码毒素和耐药性等相关基因。
Ø 病毒基因组
病毒颗粒携带的遗传物质
Ø 基因组学
所谓基因组学就是对所有基因进行作图(包括遗传图谱、物理图谱、转录图谱)、核苷酸序列分析、基因定位和基因功能分析的一门科学。简而言之,就是在基因组水平上研究基因结构和功能的科学。基因组学研究的内容包括基因的结构、组成、存在方式、表达调控模式、基因的功能及相互作用等,是研究与解读生物基因组所蕴藏的生物全部性状的所有遗传信息的一门新的前沿科学。1986年美国科学家Thomas Roderick首次提出了基因组学的概念,但随着1990年人类基因组计划启动才开始真正有系统地研究基因组、解码生命,并由“后基因组计划“的实施推动其发展。
2.基因组学的发展历程
Ø 1865年孟德尔发表豌豆杂交实验结果,提出了遗传学的两大遗传规律:分离定律、自由组合定律,并提出了“遗传因子”学说。 l
Ø 1915年美国生物学家摩尔根创立了现代遗传学的基因学说 。 l
Ø 1944年美国细菌学家艾弗里首次证明DNA是遗传信息的载体。
Ø 1953年美国生物学家沃森、英国生物物理学家克里克建立 了DNA的双螺旋结构模型,并提出了DNA的复制机制,生物学研究从此进入了分子生物学时代。
Ø 1958年克里克最初提出中心法则:DNA→RNA→蛋白质。它说明遗传信息在不同的大分子之间的转移都是单向的,不可逆的,只能从DNA到RNA(转录),从RNA到蛋白质(翻译)。
Ø 1971年美国病毒学家特明、美国病毒学家巴尔的摩发现 了“逆转录酶”,揭示了生物遗传中存在着由RNA形成DNA的过程,发展和完善了“中心法则”。 l
Ø 1983年美国生物化学家穆利斯发明利用“聚合酶链反应法 ”(PCR)。 l
Ø 1990年“人类基因组计划”正式启动,生物学研究进入基因组学时代。
Ø 2000年中、美、日、德、法、英6国科学家联合宣布成功 绘制出人类基因组草图。
Ø 2001年以后,随着功能基因组学、蛋白质组学的兴起,生物学研究进入后基因组学时代。
3.基因的结构
基因是具有遗传效应的DNA片段,无论是真核基因、原核基因都能储存、传递和表达遗传信息,也都可能发生突变,从而决定生物体的形状。基因之所以能够行使这些重要功能,与其结构有着密切的关系。那么基因的结构是什么样子的呢?真核和原核生物的基因结构是相同的吗?
3.1真核生物基因结构
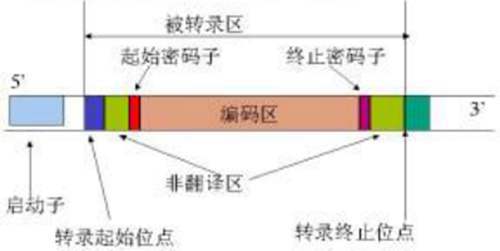
真核细胞基因结构主要由4个区域组成,即:①编码区,包括外显子与内含子;②前导区,位于编码区上游,相当于RNA 5’末端非编码区(非翻译区);③尾部区,位于RNA 3’编码区下游,相当于末端非编码区(非翻译区);④调控区,包括启动子和增强子等。
Ø 真核生物具有真正的核结构和一定数目的染色体。除配子为单倍体外,体细胞一般为二倍体。99%的DNA在核基因组(nuclear genome)中。
Ø 真核生物基因组远大于原核生物,具有多个复制起点。
Ø 大部分基因具有内含子,因此真核基因一般是不连续的,又称断裂基因。
Ø 非编码序列的量多于编码序列。
Ø 存在大量重复DNA序列:高度重复序列、中度重复顺序及单拷贝顺序等。
Ø 真核生物中未发现原核生物的操纵子结构。
① 增强子
许多真核生物启动子的转录可被远离转录起始位点数千个碱基的调控元件所增强,这一调控元件被称为增强子(enhancer)。它不能启动一个基因的转录,但有增强转录的作用。此外,增强子顺序可与特异性细胞因子结合而促进转录的进行。研究表明,增强子通常有组织特异性,这是因为不同细胞核有不同的特异因子与增强子结合,从而对基因表达有组织、器官、时间不同的调节作用。
②启动子
基因的表达是由一段位于编码序列上游的DNA调控的,这段顺序称之为启动子(promoter)。启动子中的保守序列可以被RNA聚合酶和别的与转录有关的转录因子(transcription factor)识别并结合,启动基因的RNA转录。细胞中一个基因的表达由其启动子的序列以及该启动子与RNA聚合酶和转录因子的结合能力来决定。
l 启动子是基因转录起始所必须的一段DNA序列,一般位于结构基因的上游,是DNA分子上与RNA聚合酶特异结合而使转录起始的部位,启动子本身不被转录。启动子组成如下:
Ø TATA盒(TATA Box),又称Goldberg-Hogness Box,是位于转录起点上游的一段保守序列。它的顺序为TATAAATA,位置在-34—-36之间,是决定基因转录始的选择,为RNA聚合酶的结合处之一,RNA聚合酶与TATA框牢固结合之后才能开始转录。绝大多数的真核基因都有TATA盒,TATA盒对真核基因的转录起始不是必需的,缺失仍可进行转录,但转录的起始就会不在原来的位置上,而且转录可以在若干个不同的位置上开始,产生多种转录物;同时,TATA盒中的任一碱基的突变,都引起转录的剧烈下降。因此,TATA盒决定了转录起点的正确选择,并影响转录起始的效率。在有些基因中不存在TATA盒,这样的基因中可能存在某种替代机制。
Ø CAAT盒(CAAT Box), 在某些真核基因中存在,其一致序列为GGTCAATCT。一般位于-75附近,虽然名为CAAT盒,但前面GG的重要性并不亚于CAAT部分。CAAT盒的突变会导致转录效率的急剧下降,它对某些基因的转录是必需的,对某些基因(如胸苷激酶基因)的转录则是不必要的。
Ø GC盒(GC Box),有两个拷贝,位于CAAT框的两侧,由GGCGGG组成,是一个转录调节区,有激活转录的功能。
③含子和外显子
l 大多数真核生物的基因为不连续基因(interruptesd或discontinuous gene)。所谓不连续基因就是基因的编码序列在DNA分子上是不连续的,被非编码序列隔开。编码的序列称为外显子(exon),是一个基因表达为多肽链的部分;非编码序列称为内含子(intron),又称插入序列(intervening sequence,IVS)。内含子只参与转录形成pre-mRNA,在pre-mRNA形成成熟mRNA时被剪切掉。如果一个基因有n个内含子,一般总是把基因的外显子分隔成n+1部分。内含子的核苷酸数量可比外显子多许多倍。
内含子普遍存在于真核生物和真核病毒中,在发现内含子后很长一段时间内,人们曾以为内含子是真核生物的标志。但在1983年以后,相继在原核生物中发现了内含子的存在,如大肠杆菌T4噬菌体的胸腺嘧啶核苷酸合成酶基因、硫化叶菌的亮氨酸tRNA和丝氨酸tRNA基因中发现了内含子的存在,这就打破了内含子只存在于真核生物中的概念。
每个基因中所含有的内含子的数目变化很大,可以从0到50多个不等。外显子和内含子的长度也有变化,但通常内含子比外显子要长,占了整个基因序列的大部分。内含子的特点是:5’端以GT开始, 3’以AG结束,称为GT/AG规则。
起始密码子:Kozak比较了47种植物基因与植物病毒基因中翻译起始位点附近的23个核苷酸,除了一个基因例外,其余的都是从5’端的第一个AUG作为翻译起始密码子的。例外的是菜豆的凝集素基因,它的转录本5’端有4个AUG,但彼此的读码框不同。可能是由于前面3个AUG的旁邻序列不适宜核糖体的识别而不被使用,第4个AUG是真正的翻译起始密码子。
④终止子
l 终止子在一个基因的末端往往有一段特定顺序,它具有转录终止的功能,这段终止信号的顺序称为终止子(termianator)。终止子的共同顺序特征是在转录终止点之前有一段回文顺序,约7-20核苷酸对。回文顺序的两个重复部分分由几个不重复碱基对的不重复节段隔开,回文顺序的对称轴一般距转录终止点16-24bp。
③ 5’非翻译区
基因的转录起始位点到翻译起始密码子之间的一段序列被称为5’非翻译区。该区的5’端是前体mRNA加帽(7-甲基鸟嘌呤核苷)的位点。有些基因的5’非翻译区中还有一些茎环结构,这些茎环结构与翻译起始密码子中的旁邻序列对翻译的效率都有影响。此外有些植物基因的5’非翻译区中还鉴定出有内含子存在。如在Ubiquitin基因、Sh基因、Actin基因、Adhl基因与Wx基因中的5’非翻译区中均有内含子存在,且这些内含子都有增强基因表达的作用。
3.2原核生物基因结构
原核生物基因分为编码区与非编码区。编码区就是能转录为相应的信使RNA,进而指导蛋白质的合成,也就是说能够编码蛋白质。非编码区则相反,但是非编码区对遗传信息的表达是必不可少的,因为在非编码区上有调控遗传信息表达的核苷酸序列。非编码区位于编码区的上游及下游。在调控遗传信息表达的核苷酸序列中最重要的是位于编码区上游的RNA聚合酶结合位点。RNA聚合酶是催化DNA转录为RNA,能识别调控序列中的结合位点,并与其结合。
原核生物的基因结构特点 :①为一条环状双链DNA;②只有一个复制起点;③具有操纵子结构;④绝大部分为单拷贝;⑤基因一般是连续的,无内含子;⑥基因组含有大量单一序列,仅有少量的重复顺序和基因。
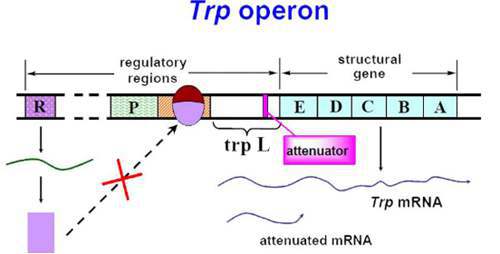
操纵子(operon)是指包含结构基因、操纵基因以及启动基因的一些相邻基因组成的DNA片段,其中结构基因的表达受到操纵基因的调控。主要见于原核生物,但在真核生物中也存在。
原核生物大多数基因表达调控是通过操纵子机制实现的。操纵子通常由 2个以上的编码序列与启动序列、操纵序列以及其他调节序列在基因组中成簇串联组成。启动序列是RNA聚合酶结合并启动转录的特异DNA序列。多种原核基因启动序列特定区域内,通常在转录起始点上游-10及-35区域存在一些相似序列,称为共有序列。操纵序列是原核阻遏蛋白的结合位点。当操纵序列结合阻遏蛋白时会阻碍RNA聚合酶与启动序列的结合,或使RNA聚合酶不能沿DNA向前移动,阻遏转录,介导负性调节。原核操纵子调节序列中还有一种特异DNA序列可结合激活蛋白,使转录激活,介导正性调节。
原核生物大多数基因表达调控是通过操纵子机制实现的,如乳糖操纵子、阿拉伯糖操纵子、组氨酸操纵子、色氨酸操纵子等。
小结:真核生物和原核生物的基因在结构上的主要区别是?
区别:原核生物的DNA的编码区是连续的,真核生物DNA的编码区是间断的,即真核生物的DNA的编码区有内含子和外显子,复制时同时复制内含子和外显子,翻译的最终结果是成熟的mRNA只携带外显子的遗传信息,内含子的遗传信息被切除。原核生物的DNA的编码区没有内含子和外显子一说,全部是有遗传意义的片断,是完全翻译。另外,真核细胞的基因完全是由DNA构成,而有的原核基因还会由RNA构成,例如RNA病毒。
相同点:都有编码区和非编码区,在非编码区都有调控遗传信息表达的核苷酸序列,在编码区上游都有与RNA聚合酶结合位点(启动子)。
3.3基因组变异类型
基因的遗传变异主要分为两类:可遗传的变异和不可遗传的变异。可遗传变异是生物体内遗传物质发生变化而造成的一种可以遗传给后代的编译,正是这种变异导致了生物在不同水平上体现出遗传多样性。;不可遗传的变异是由环境引起的,遗传物质没有发生变化。可遗传的变异的来源主要有3个:基因重组、基因突变和染色体变异。
基因组的遗传变异类型多样,囊括了单核苷酸多态性(SNP),小片段的插入和缺失(Indel),结构变异(SV),拷贝数变异(CNV)等。
Ø 单核苷酸多态性(SNP)
单核苷酸多态性(Single Nucleotide Polymorphism,SNP)主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。它是人类可遗传的变异中最常见的一种, 占所有已知多态性的80%以上。SNP在人类 基因组中广泛存在,平均每500〜1000个碱基 对中就有1个,估计其总数可达300万个甚至更多。SNP是一种二态的标记,由单个碱基的转换或颠换所引起,也可由碱基的插入或缺失所致,但通常所说的SNP并不包括后两种情况。SNP既可能在基因序列内,也可能在基因以外的非编码序列上。
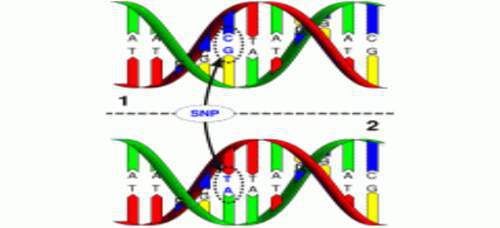
理论上讲,SNP既可能是二等位多态性,也可能是3个或4个等位多态性,但实际上,后两者非常少见,几乎可以忽略。因此,通常所说的SNP都是二等位多态性的。这种变异可能是转换(C←→T,在其互补链上则为G←→A),也可能是颠换(C←→A,G←→T,C←→G,A←→T)。转换的发生率总是明显高于其它几种变异,具有转换型变异的SNP约占2/3,其它几种变异的发生几率相似。研究证明了,转换的几率之所以高,可能是因为CpG二核苷酸上的胞嘧啶残基是基因组中最易发生突变的位点,其中大多数是甲基化的,可自发地脱去氨基而形成胸腺嘧啶。
在遗传学分析中, SNP 作为一类遗传标记得以广泛应用, 主要源于这几个特点:
(1)密度高:SNP在人类基因组的平均密度估计为 1\1000 bp , 在整个基因组的分布达 3×106个,遗传距离为 2~3cM , 密度比微卫星标记更高, 可以在任何一个待研究基因的内部或附近提供一系列标记。
(2)富有代表性:某些位于基因内部的SNP 有可能直接影响蛋白质结构或表达水平, 因此, 它们可能代表疾病遗传机理中的某些作用因素。SNP自身的特性决定了它更适合于对复杂性状与疾病的遗传解剖以及基于群体的基因识别等方面的研究。
(3)遗传稳定性:与微卫星等重复序列多态性标记相比, SNP 具有更高的遗传稳定性。
(4)易实现分析的自动化:SNP标记在人群中只有两种等位型(allele) 。这样在检测时只需一个“ + \- ”或“全\无”的方式,而无须象检测限制性片段长度多态性,微卫星那样对片段的长度作出测量,这使得基于SNP的检测分析方法易实现自动化。
Ø 插入缺失(Indel)
InDel (insertion-deletion) 插入缺失标记,指的是同一物种不同个体间,在同一位点的DNA序列处发生了核苷酸片段的插入或者缺失,长度通常在50bp以内,在该位点两侧设计特异引物进行PCR扩增,不同个扩增出来的片段大小会存在差异。
Ø 结构变异(Structure Variation,SV)
(染色体)结构变异,包括插入、缺失、倒位、易位及重复等,长度在1kb~3Mb。
Ø 拷贝数变异(CNV)
拷贝数变异(Copy number variation, CNV)是由基因组发生重排而导致的, 一般指长度为1 kb ~3kb的基因组大片段的拷贝数增加或者减少, 主要表现为亚显微水平的缺失和重复。CNV 是基因组结构变异(Structuralvariation, SV) 的重要组成部分。CNV位点的突变率远高于SNP(Single nucleotide polymorphism), 是人类疾病的重要致病因素之一。
目前, 用来进行全基因组范围的 CNV 研究的方法有: 基于芯片的比较基因组杂交技术(array-based comparative genomic hybridization, aCGH)、SNP 分型芯片技术和新一代测序技术。CNV的形成机制有多种, 并可分为DNA重组和DNA错误复制两大类。CNV可以导致呈孟德尔遗传的单基因病与罕见疾病, 同时与复杂疾病也相关。其致病的可能机制有基因剂量效应、基因断裂、基因融合和位置效应等。对 CNV的深入研究, 可以使我们对人类基因组的构成、个体间的遗传差异、以及遗传致病因素有新的认识。
3.4基因组变异检测方法
目前主要有4种检测基因组上结构性变异的策略,这四种方法的核心思路都是将序列和参考基因组比对,基于已知信息查找变异信号。这四种策略分别为:(1)Read pair(也称为Pair-end Mapping,简称PEM);(2)Split read(简称SR);(3)Read Depth(简称RD)和(4)基于de novo组装的方法。同时生物信息研究人员也已开发了众多根据以上4中策略中一种或者多种的软件用于结构性变异的检测。
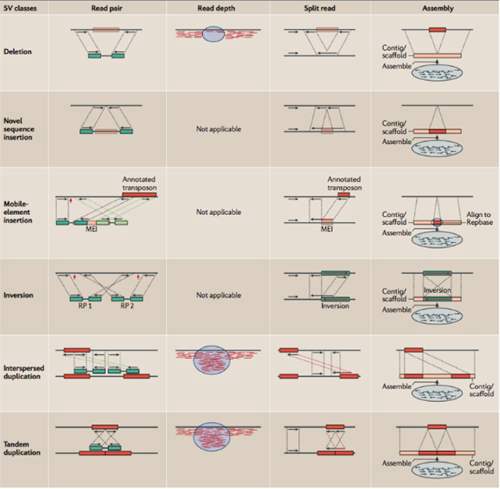
1. 基于Pair-end Mapping(PEM)
理论上来讲,PEM方法能够检测到的变异类型包括:序列删除(deletion),序列插入(insertion),序列转置(inversion),染色体内部和染色体外部的易位(intra- and inter-chromosome translocation),序列串联倍增(tandem duplications)和序列在基因组上的散在倍增(interspersed duplications)。这里有两个地方需要指出,第一,对于序列删除的检测,其所能检测到的片段长度受插入片段长度的标准差(SD)所影响(这里的插入片段长度指的是测序之前在构建DNA测序文库阶段,所选取的经由超声波打断的DNA片段长度,这些片段也称之为测序片段,这是实验过程中的操作,并不是指基因组的变异),并且越大的序列删除约容易被检测到,并且准确性也越高;第二,其所能检测的序列插入,长度只能在插入片段长度的范围内,并且最大长度也受限于测序的插入片段长度的标准差。目前,Breakdancer是应用PEM方法的软件,也是在使用变异检测方面用得最广泛的软件之一。其他类似的软件还包括:VariationHunter10, Spanner, PEMer11等等。但是,事实上整个过程并不像流程图中看起来的那么简单,而且绝大多数的软件都在检测复杂的序列结构方面(如序列易位和序列倍增)存在很大的困难。
2. Split Read(分裂read,简称SR)
对于这个方法,首先要求比对软件具备soft-clip reads的能力,如BWA 比对软件。目前illumina测序平台Pair-End测序的方法是对测序片段的两端来进行的,所以每次获得的都是来自同一个测序序列片段两端的一对read。当BWA成功地将这一对reads中的一条比对到参考序列上,而另一条却无法正常比上的时候,BWA会对这条read没能正常比上的read尝试在比对上的那条read附近使用更为宽松的Smith-Waterman局部比对策略搜索可能的比对位置。如果这条read只有一部分能够比上,那么BWA会对其进行soft-clip,而这里也往往是包含结构性变异的断点之处。Pindel12,这是目前唯一一个使用SR方法进行变异检测的软件。它在千人基因组计划和生物信息分析人员中被广泛使用。图1中也清楚地展示了Split reads的信号如何被用来进行结构性变异的检测。首先,在获得了单端唯一比对到基因组上的PE read之后,Pindel会将不能比上的那条read切开成2或者3小段,然后再分别重新按照用户所设置的最大序列删除长度去比对,并获得最终的比对位置和比对方向,而断点位置的确定则是根据soft-clipped的结果来获得。
Pindel 理论上能够检测所有长度范围内的deletion,和小片段的insertion(长度在50bp以下),inversion,tandem duplication和一些large insertion。不过目前,作者并未公开发布关于检测lager insertion的原理。Split-reads的一个优势就在于,它们精确到单碱基。但是也和大多数的PEM方法一样,Pindel同样无法解决复杂结构性变异的情形。
3. Read Depth (read 覆盖深度,简称RD)
目前存在两种利用Read depth的信息检测大拷贝数变异(Copy number variation,包括丢失序列和序列重复倍增,简称CNV)的策略。一种是,通过检测样本在一个参考基因组上read的深度分布情况来检测CNV,适用于单样本;另一种则是通过和识别出比较两个样本中所存在的丢失和重复倍增区,以此来获得相对的CNV,适用于case-control模型的样本。这有点像CGH芯片。CNVnator使用的是第一种策略,同时也广泛地被用于检测大的CNV。当然还有一些比较冷门的软件,但是由于他们没有发表相应的文章,这里就不再列举了。CNV-seq使用的是第二个策略。基于其原理,RD的方法能够很好地用于检测一些大的deletion或者duplication事件,但是对于小的变异事件就无能为力了。
4. 基于De novo assembly
理论上来讲,de novo assembly 的方法应该要算是基因组变异检测上最有效的方法了。就目前来说,它能够提供(特别是)对于long insertion和复杂结构性变异的最好检测方法。现在虽然研究人员开发了很多基于第二代测序技术数据来进行组装的软件,但是组装却仍然是一件棘手的事情,特别是脊椎动物的组装则更是如此。其中最主要的原因在于,脊椎动物基因组上所存在的重复性序列和序列的杂合会严重影响组装的质量,除去资金成本,这也在很大程度上阻碍了利用组装的方法在基因组变异检测方面的应用。
5.小结
通过对上面四种不同的变异检测策略的比较可以发现,小长度范围内的变异以及较长的deletion,目前都能够较好地检测出来,但对于大多数的long insertion和更复杂的结构性变异情况,当前的检测软件基本都没法还解决。Assembly应是当前全面获得基因组上各种变异的最好方法,但是目前的局限却也发生在Assembly本身,若是基因组没能装得好,后面的变异检测就更是无从说起。从目前的情况看,de novo assembly的方法并不能很快进入实际的应用。因此,暂且不提assembly,其余的三种策略都各有各的优势,从目前的结果看,并没有哪一款软件能够一次性地将基因组上的各种不同情况变异类型都获得。因此就目前短reads高通量测序技术来说,最合适的方案应是结合多个不同的策略,将结果合并在一起,这样可以最大限度地将FP降低。HugeSeq pipeline在这方面做了一个比较好的总结,这个软件整合了BreakDancer, CNVnator, Pindel,BreakSeq以及GATK的结果。能够给出一个相对比较准确的变异检测结果。
4.基因的表达——中心法则
中心法则(Genetic Central Dogma)是指遗传信息从DNA传递给RNA,再从RNA传递给蛋白质,即完成遗传信息的转录和翻译的过程。也可以从DNA传递给DNA,即完成DNA的复制过程。这是所有有细胞结构的生物所遵循的法则。在某些病毒中的RNA自我复制(如烟草花叶病毒等)和在某些病毒中能以RNA为模板逆转录成DNA的过程(某些致癌病毒)是对中心法则的补充。
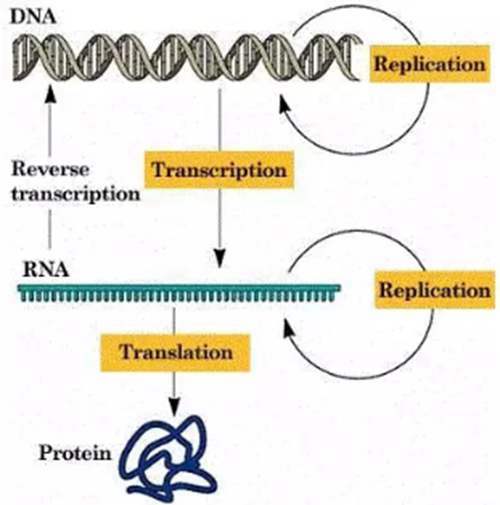
由此可见,遗传信息并不一定是从DNA单向地流向RNA,RNA携带的遗传信息同样也可以流向DNA。但是DNA和RNA中包含的遗传信息只是单向地流向蛋白质,迄今为止还没有发现蛋白质的信息逆向地流向核酸。这种遗传信息的流向,就是中心法则的遗传学意义。
小故事:病原体朊粒(Prion)的行为曾对中心法则提出了严重的挑战。是什么原因呢?
朊粒是一种蛋白质传染颗粒(proteinaceous infectious particle),是羊瘙痒病的病原体,可在羊群中传染,意味着这种病原体是能在宿主动物体内自行复制的感染因子。朊粒同时又是人类的中枢神经系统退化性疾病的病原体,也可引起疯牛病。研究证明,这种朊粒不是病毒,而是不含核酸的蛋白质颗粒!!!
那么问题来了:一个不含DNA或RNA的蛋白质分子能在受感染的宿主细胞内产生与自身相同的分子,且实现相同的生物学功能,即引起相同的疾病,是不是意味着这种蛋白质分子也是负载和传递遗传信息的物质?如果情况属实,这将从根本上动摇遗传学基础!!!
后期实验证明,朊粒确实是不含DNA和RNA的蛋白质颗粒,但它不是传递遗传信息的载体,也不能自我复制,而仍是由基因编码产生的一种正常蛋白质的异构体,它的复制方式是朊粒(SC型PrP型蛋白)接触到了生物体内正常的C型PrP蛋白,导致C型的变成了SC型,多么神奇的存在~
关于朊粒的传说:其实,我是bug —— 朊病毒的自传
小知识:人类基因组约含有30亿个碱基,需要3G的储存空间。将人体全部细胞中的DNA展开,是来回月球距离的6000倍。人与人之间的DNA序列99.9%是相同的,只有0.1%的差异。人类与黑猩猩的DNA有94%~99%相同,有研究表明,人类至少有1~4%的穴居人DNA,也许在某个时候智人和穴居人相互混居。
4.1 DNA→DNA(复制)
DNA是分子结构复杂的有机化合物,作为染色体的一个组分而存在于细胞核内,功能为储藏遗传信息,是遗传的主要物质基础,是遗传信息的贮存场所。DNA的组成单位为4种脱氧核苷酸:腺嘌呤脱氧核苷酸(dAMP )、胸腺嘧啶脱氧核苷酸(dTMP )、胞嘧啶脱氧核苷酸(dCMP )及鸟嘌呤脱氧核苷酸(dGMP ),这4种核苷酸可以任何次序排列连接成数百万个核苷酸长链聚合物分子,以双螺旋的结构存在于生物体中。
DNA的复制是遗传物质从亲代转移至子代的关键,DNA为双链结构,其复制是一个边解旋边复制的过程,称为半保留复制,因为通过复制所形成的新的DNA分子,保留原来亲本DNA双链分子的一条单链。复制的过程主要包括:起始,延伸,终止。需要的环境条件主要包括:
模板:DNA的两条链
原料:dATP、dTTP、dCTP、dGTP
酶:DNA聚合酶、解旋酶、DNA连接酶等
能量:ATP高能磷酸键
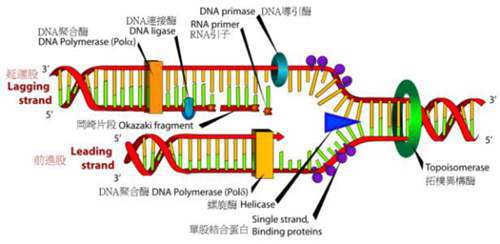
① 复制起始
在该阶段首先DNA复制起始位点被DNA解旋酶解开形成复制叉,第二步RNA聚合酶沿滞后链模版转录一短的RNA分子,第三步前导链DNA模版上开始合成RNA引物随后DNA聚合酶将第一个脱氧核苷酸加到引物RNA的3-OH末端复制引发的关键步骤就是前导链DNA的合成,一旦前导链DNA的聚合作用开始,滞后链上的DNA合成也随着开始。
在这个阶段需要的蛋白有:
拓扑异构酶:帮助解开复制叉前后的超螺旋结构
DNA解旋酶:解开螺旋
Rep蛋白:帮助解开双螺旋结构
单链结合蛋白:稳定单链区
引物合成酶:合成RNA引物
DNA聚合酶I:消除引物,填满裂缝
DNA聚合酶III:合成DNA
DNA连接酶:连接DNA末端
需要值得注意的是:
不论是前导链还是后随链,都需要一段RNA引物用于开始子链DNA合成。因此前导链和后随链的差别在于前者从复制起始点开始按5’—3’持续的合成下去、不形成冈崎片段、后者则随着复制叉的出现、不断合成长约2—3kb的冈崎片段。
为什么需要有RNA引物来引发DNA复制呢?这可能尽量减少DNA复制起始处的突变有关。DNA复制开始处的几个核苷酸最容易出现差错,因此,用RNA引物即使出现差错最后也要被DNA聚合酶I切除,提高了DNA复制的准确性。
② 延伸阶段
进入此阶段的标志是DNA聚合酶III把第一个核苷酸加到引物3‘-OH端。在此阶段,DNA解旋酶在复制叉处边移动边解开DNA模版双链,复制体在DNA前导链模板和滞后链模板上移动并合成了连续的DNA前导链和由许多冈崎片段组成的滞后链。在DNA前导链合成延伸过程中主要是DNA聚合酶Ⅲ的作用。当冈崎片段形成后,DNA聚合酶Ⅰ通过其5→3外切酶活性切除冈崎片段上的RNA引物,同时,利用后一个冈崎片段作为引物由5→3合成DNA。最后两个冈崎片段由DNA连接酶将其接起来,形成完整的DNA滞后链。随着解旋过程的进行,新合成的子链也不断地延伸,同时,每条子链与其对应的母链盘绕成双螺旋结构,从而各形成一个新的DNA分子。
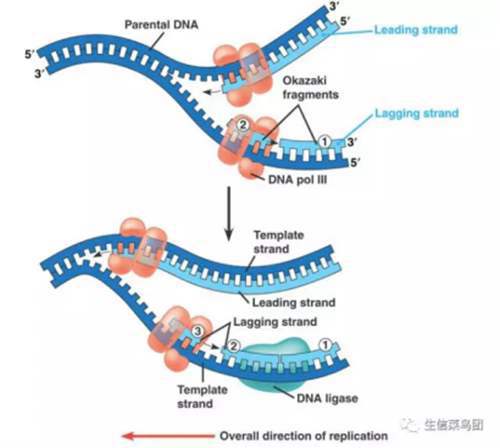
在这个阶段需要的蛋白有:
DNA聚合酶III:合成DNA
引发体:由引发前体(与引发蛋白)和引发酶组成,引发体能够在后随链模板上移动,移动过程需ATP供能,移动方向与后随链合成方向相反(沿着后随链模板5’→3’方向移动),且与复制叉的移动保持同步
DNA聚合酶I:通过其5→3外切酶活性切除冈崎片段上的RNA引物,同时,利用后一个冈崎片段作为引物由5→3合成DNA。
DNA连接酶:连接两个岗崎片段
③ 终止阶段
过去认为,DNA一旦复制开始,就会将该DNA分子全部复制完毕才终止其DNA复制。但最近的实验表明,在DNA上也存在着复制终止位点,DNA复制将在复制终止位点处终止,并不一定等全部DNA合成完毕。但对复制终止位点的结构和功能了解甚少。如已有研究证明大肠杆菌染色体DNA具有复制终止位点,此处可以结合一种特异的蛋白质分子叫做Tus,这个蛋白质可能是通过阻止解链酶(Helicase)的解链活性而终止复制的。详细的机制还不完全清楚。
DNA复制完成后,靠拓扑酶将DNA分子引入超螺旋结构。
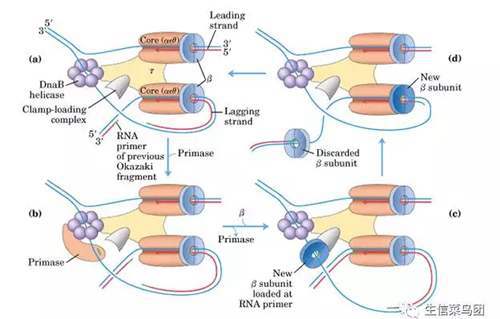
DNA复制的特点:
半保留复制:DNA在复制时,以亲代DNA的每一股作模板,合成完全相同的两个双链子代DNA,每个子代DNA中都含有一股亲代DNA链
有一定的起始位点:DNA在复制时,需在特定的位点起始,这是一些具有特定核苷酸排列顺序的片段,即复制起始点(复制子)。在原核生物中,复制起始点通常为一个,而在真核生物中则为多个。
需要引物:DNA聚合酶必须以一段具有3端自由羟基(3-OH)的RNA作为引物,才能开始聚合子代DNA链。RNA引物的大小,在原核生物中通常为50~100个核苷酸,而在真核生物中约为10个核苷酸。
双向复制:DNA复制时,以复制起始点为中心,向两个方向进行复制。但在低等生物中,也可进行单向复制。
半不连续复制:由于DNA聚合酶只能以5→3方向聚合子代DNA链,因此两条亲代DNA链作为模板聚合子代DNA链时的方式是不同的。
4.2 DNA→RNA(转录)
转录是指遗传信息从基因(DNA)转移到RNA,即信使RNA(mRNA)以及非编码RNA(tRNA、rRNA等)的合成步骤。
转录中,一个基因会被读取、复制为mRNA;这个过程由RNA聚合酶(RNA polymerase)和转录因子(transcription factor)所共同完成。
RNA聚合酶通过与一系列组分构成动态复合体,完成转录起始、延伸、终止等过程。生成的mRNA携有的密码子,进入核糖体后可以实现蛋白质的合成。转录仅以DNA的一条链作为模板,被选为模板的单链称为模板链,亦称无义链;另一条单链称为非模板链,即编码链,因编码链与转录生成的RNA序列一致,所以又称有义链。
RNA的合成一般分两步,第一步合成原始转录产物(过程包括转录的启动、延伸和终止);第二步转录产物的后加工,使无生物活性的原始转录产物转变成有生物功能的成熟RNA。PS:原核生物mRNA的原始转录产物一般不需后加工就能直接作为翻译蛋白质的模板。
① 启动
RNA聚合酶识别DNA编码链上的启动子并形成由酶、DNA和核苷三磷酸(NTP)构成的三元起始复合物,转录即自此开始。第一个核苷三磷酸与第二个核苷三磷酸缩合生成3′-5′磷酸二酯键后,则启动阶段结束,进入延伸阶段。DNA模板上的启动区域常含有TATAATG顺序,称TATA盒。
② 延伸
σ亚基脱离酶分子,留下的核心酶与DNA的结合变松,因而较容易继续往前移动。核心酶无模板专一性,能转录模板上的任何顺序。脱离核心酶的σ亚基还可与另外的核心酶结合,参与另一转录过程。随着转录不断延伸,DNA双链顺次地被打开,并接受新来的碱基配对,合成新的磷酸二酯键后,核心酶向前移去,已使用过的模板重新关闭起来,恢复原来的双链结构。
③ 终止
转录的终止包括停止延伸及释放RNA聚合酶和合成的RNA。在原核生物基因或操纵子的末端通常有一段终止序列即终止子;RNA合成就在这里终止。原核细胞转录终止需要一种终止因子ρ(四个亚基构成的蛋白质)的帮助。真核生物DNA上也可能有转录终止的信号。已知真核DNA转录单元的3′端均含富有AT的序列〔如AATAA(A)或ATTAA(A)等〕,在相隔0~30bp之后又出现TTTT顺序(通常是3~5个T),这些结构可能与转录终止或者与3′端添加多聚A顺序有关。
④ RNA概述及分类
基因只占人类基因组的3%,其余97%都是非编码序列。非编码序列也是可以表达的,其表达产物就是非编码RNA。人类基因组中约93%的DNA是能转录为RNA的,其中2%是mRNA,98%是非编码RNA(ncRNA)。非编码RNA(ncRNA)可以分为调控RNA和管家RNA两种。
调控RNA
miRNA:微RNA (microRNA),18-25 nt.,单链。
siRNA:小干扰RNA (smallinterfering RNA),21-23 nt.,双链。
piRNA:piwi相互作用RNA (piwi-interacting RNA),26-35 nt.,单链,这是动物生殖细胞所特有的小RNA,转座子沉默。
lncRNA:长非编码RNA (long non-coding RNA),500 nt.,比如Xist、PCGEM1等。
管家RNA
rRNA:核糖体RNA (ribosome RNA),26-35 nt.,单链,是构成核糖体的组成成分,有多种不同的大小,如28S、18S、5S等等。
tRNA:转运RNA (transfer RNA),70-80 nt.,单链,三叶草构型,在蛋白质合成过程中起到转运氨基酸的作用。对于不同的物种,其rRNA分子的大小和种类都可能有所不同。
snoRNA:核仁小RNA (smallnucleolar RNA)
sacRNA:Small Cajal body-specific RNAs,是一种特殊的核仁小RNA,专一位于卡哈尔体(Cajal body)上,可以催化核糖核蛋白的生成。
Telomerase RNA:端粒酶RNA,是端粒酶的一部分,在端粒延伸过程中,作为端粒继续延伸的模板,由端粒酶催化实现端粒的延长。
热门ncRNA——lncRNA、miRNA、circRNA
目前研究最热门的ncRNA主要集中在lncRNA、miRNA、circRNA三种。
IncRNA
lncRNA可通过折叠形成一定的空间结构与多种蛋白互作,也可通过碱基互补配对与其它核酸进行识别,这种识别又可将蛋白引导至特定序列位点,这些特点使得lncRNA在发育和癌症中的功能发挥得更加丰富。
作为RNA诱饵,结合转录因子,干扰其与基因promoter区域的结合,从而调控转录;
作为分子海绵,吸附miRNA,抑制其与mRNA的结合,使得mRNA免于降解;
作为蛋白互作的支架或桥梁,影响蛋白多聚物的形成,调控蛋白活性;
招募染色质修饰因子,改变染色质的修饰水平,从而影响基因的转录和表达;
与mRNA配对结合,抑制翻译;
与mRNA配对结合,影响剪切;
与mRNA配对结合,影响mRNA的稳定性。
circRNA
circRNA分子呈封闭环状结构,无游离5‘和3’末端,不易被核酸外切酶RNaseR降解,比线性RNA更加稳定。 长度约200-2000bp,主要长度分布在500bp左右。
circRNA大多数来源于外显子,少部分由内含子直接环化形成。其形成有四种模式,a:套索驱动的环化 ;b:内含子碱基配对驱动环化;c:单个内含子成环 ;d:RNA结合蛋白驱动环化。
它可以通过竞争性结合miRNA、线性亲本基因的转录,甚至是编码多肽来发挥生物学功能。
A. circRNA作为ceRNA(内源竞争性RNA)竞争性结合miRNA;
B. circRNA结合RNA结合蛋白(RBP)以形成RNA-蛋白复合物(RPC),调控线性亲本基因的转录;
C. 编码功能,circRNA具有内部核糖体进入位点(IRES),能合成多肽。
miRNA
miRNA一类由内源基因编码的非编码单链RNA分子,其长度约为19-25nt,其在肿瘤发生发展、生物发育、器官形成、病毒防御、表观调控以及代谢等方面起着极其重要的调控作用。
RNA聚合酶II/III转录成pri-miRNA,Drosa/DGCR8复合体将其裂解为pre-miRNA(前体miRNA);
Exportin-5-Ran-GTP复合物将pre-miRNA转运出核;
Dicer酶裂解pre-miRNA至成熟的长度(19-25nt);
双链的miRNA被转载进AGO2,一条链降解,一条链形成RISC,发挥生物学功能。
生物学功能有:mRNA的裂解及降解、抑制翻译。此外还有转录调控功能。
以上就是(基因组学一)全部内容,收藏起来下次访问不迷路!